Search Results
184 results found with an empty search
- Berberine: A Natural Anti-Parasitic Agent
Berberine Anti-Parasitic Animated Intro for Gut Health and Microbial Balance Berberine is a bright yellow plant alkaloid found in several medicinal herbs including: goldenseal barberry Oregon grape tree turmeric For centuries, traditional medical systems have used berberine-containing plants for digestive illness, diarrhea, gastrointestinal infections, and inflammatory disorders. Today, berberine is widely discussed for: blood sugar support insulin resistance metabolic syndrome cholesterol reduction However, one of its oldest traditional uses involved intestinal infections and parasitic illness. Modern laboratory studies now suggest berberine possesses broad antimicrobial properties that may include anti-parasitic activity. What Is a Parasite? Understanding Intestinal Parasites and Berberine Support Parasites are organisms that survive by living within or on another organism. Intestinal parasites may include: protozoa amoebae helminths (worms) giardia blastocystis species cryptosporidium Symptoms may include: bloating diarrhea abdominal pain nausea fatigue malabsorption weight changes nutrient deficiencies Some infections are mild while others can become serious, particularly in immunocompromised individuals. How Might Berberine Work Against Parasites? Berberine appears to work through several mechanisms. 1. Direct Antimicrobial Activity Berberine has demonstrated activity against: bacteria fungi yeast protozoa Laboratory studies suggest berberine may interfere with microbial replication, energy metabolism, and cellular membrane stability.^1^ Some studies suggest activity against: Giardia lamblia Entamoeba histolytica Blastocystis hominis Candida species 2. Disruption of Biofilms Many microorganisms protect themselves within biofilms — protective layers that make infections harder to eradicate. Berberine may help interfere with biofilm formation, potentially improving microbial clearance.^2^ 3. Reduction of Intestinal Inflammation Parasitic infections often trigger intestinal inflammation and disruption of the gut barrier. Berberine appears to possess: anti-inflammatory effects antioxidant properties intestinal barrier support This may help reduce: bloating intestinal irritation inflammatory signaling How Berberine Supports Gut Health and Anti-Parasitic Defense Berberine and Gut Microbiome Balance One of the more interesting aspects of berberine is that it may help shift gut microbial balance rather than simply acting as a broad “gut sterilizer.” Research suggests berberine may influence: microbial diversity inflammatory pathways short-chain fatty acid production metabolic signaling This may partially explain why berberine is also studied in: insulin resistance obesity metabolic syndrome fatty liver disease Berberine Compared with Conventional Therapy Some parasitic infections require prescription medications and physician-directed treatment. Patients with these symptoms, should undergo proper medical evaluation: persistent diarrhea blood in stool dehydration fever severe abdominal pain unexplained weight loss Berberine should not be viewed as a replacement for appropriate diagnosis or treatment of serious infections. However, in integrative medicine, berberine is sometimes used as part of broader gastrointestinal support programs under physician supervision. Berberine Integrative Gut Health and Anti-Parasitic Care Typical Dosing Most berberine supplements range from: 400–600 mg taken 2–3 times daily Because berberine can affect blood sugar and metabolism, patients should discuss supplementation with their healthcare provider. Possible Side Effects Berberine is generally well tolerated but may cause: constipation stomach upset cramping nausea diarrhea Berberine may also interact with: diabetes medications anticoagulants blood pressure medications cyclosporine some antibiotics Berberine should generally be avoided during pregnancy unless specifically directed by a physician. The Importance of Proper Diagnosis Not all gastrointestinal symptoms are caused by parasites. Other possible causes include: bacterial overgrowth food intolerance inflammatory bowel disease pancreatic insufficiency fungal overgrowth metabolic disease Modern stool PCR testing and gastrointestinal diagnostic panels may help identify underlying contributors more accurately than traditional methods alone. Related Topics Insulin Resistance: The Hidden Precursor to Cardiovascular Disease, Dementia, and Accelerated Aging Metabolic Syndrome: The Clinical Turning Point Sepsis: What Is It and How Does It Present? Risks of Untreated Bacteria in the Urine Kombucha and Gut Health Cacao and Cardiovascular Health Bottom Line Berberine is a fascinating natural compound with growing evidence supporting antimicrobial, anti-inflammatory, metabolic, and possible anti-parasitic effects. While it should not replace appropriate medical diagnosis and treatment, berberine may provide useful support within a broader integrative gastrointestinal strategy. As interest in gut health continues to grow, berberine remains one of the most extensively studied botanical compounds in functional and integrative medicine. Become a Patient At Stages of Life Medical Institute, we provide comprehensive evaluations focused on gastrointestinal health, metabolic wellness, inflammation reduction, integrative medicine, and preventive care. References Birdsall TC, Kelly GS. Berberine: therapeutic potential of an alkaloid found in several medicinal plants. Altern Med Rev. 1997;2(2):94-103. PMID: 9258793. Habtemariam S. Berberine pharmacology and the gut microbiota. Biomed Pharmacother. 2020;131:110647. PMID: 32827909. Imenshahidi M, Hosseinzadeh H. Berberis vulgaris and berberine: an update review. Phytother Res. 2016;30(11):1745-1764. PMID: 27573202. PubMed Reference 3 Tillhon M, et al. Berberine: new perspectives for old remedies. Biochem Pharmacol. 2012;84(10):1260-1267. PMID: 22940211. PubMed Reference 4 Zhang X, et al. Berberine and gut microbiota modulation. Front Cell Infect Microbiol. 2020;10:504. PMID: 33014802. PubMed Reference 5 Cicero AFG, Baggioni A. Berberine and metabolic disorders. Nutrients. 2016;8(5):269. PMID: 27164058. PubMed Reference 6 Feng X, Sureda A, et al. Berberine in cardiovascular and metabolic disease. Oxid Med Cell Longev. 2019;2019:2501426. PMID: 31467642. Wang Y, et al. Berberine and intestinal barrier function. Pharmacol Res. 2021;165:105459. PMID: 33359612. PubMed Reference 8 Ortiz LM, et al. Antimicrobial mechanisms of berberine. Clin Microbiol Rev. 2014;27(3):647-679. PMID: 24982372. PubMed Reference 9 Kong WJ, et al. Berberine and gastrointestinal health. World J Gastroenterol. 2015;21(27):8786-8793. PMID: 26217066. PubMed Reference 10 REFERENCES The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Bergamot Extract and Cholesterol Lowering: Natural Support for Heart Health
Bergamot Extract for Cholesterol Lowering and Cardiovascular Health Bergamot is a fragrant citrus fruit primarily grown in southern Italy. Bergamot is a fragrant citrus fruit primarily grown in southern Italy. While many people recognize bergamot as the distinctive flavoring in Earl Grey tea, researchers have increasingly focused on bergamot extract for its potential cardiovascular and metabolic benefits. Earl Grey tea, researchers have increasingly focused on bergamot extract for its potential cardiovascular and metabolic benefits. Bergamot extract has become one of the most discussed natural supplements in integrative cardiovascular medicine. Derived from Citrus bergamia, bergamot extract contains concentrated polyphenols and flavonoids that may support healthy cholesterol levels, vascular health, and metabolic function. Unlike the culinary fruit itself, the supplements studied in clinical research are typically highly concentrated bergamot polyphenol extracts standardized for active compounds associated with lipid metabolism and antioxidant activity. For patients seeking additional natural support for cardiovascular health, bergamot extract has generated increasing scientific interest. What Is Bergamot Extract, and How Does It Lower Cholesterol? Bergamot extract is produced primarily from the juice and peel fractions of bergamot citrus grown in southern Italy. The extract contains several bioactive compounds including: brutieridin melitidin neoeriocitrin naringin neohesperidin These polyphenols appear to influence: LDL cholesterol metabolism triglyceride production oxidative stress vascular inflammation endothelial function insulin sensitivity Bergamot Extract Benefits for Cholesterol and Heart Health How Might Bergamot Extract Lower Cholesterol? 1. LDL Cholesterol Reduction Low-density lipoprotein (LDL) cholesterol contributes to arterial plaque formation and cardiovascular disease risk. Several bergamot polyphenols appear to influence HMG-CoA reductase pathways involved in cholesterol synthesis.^1^ This mechanism resembles part of the pathway targeted by statin medications, although bergamot extract should not be viewed as a direct replacement for prescription therapy. Clinical studies have demonstrated meaningful reductions in LDL cholesterol in some patients using bergamot extract supplementation.^2^ 2. Triglyceride Reduction Elevated triglycerides are strongly associated with: insulin resistance obesity metabolic syndrome fatty liver disease cardiovascular inflammation Research suggests bergamot extract may help reduce triglyceride levels while improving lipid balance.^3^ 3. HDL Cholesterol Support Some studies suggest bergamot extract may modestly improve HDL (“good cholesterol”) levels while improving the LDL-to-HDL ratio. Improving overall lipid balance may be as important as lowering LDL alone. 4. Antioxidant and Anti-Inflammatory Effects Oxidative stress damages blood vessels and contributes to plaque instability. Bergamot extract contains powerful antioxidant polyphenols that may help: reduce oxidized LDL improve endothelial function support nitric oxide activity decrease inflammatory signaling This may support overall vascular resilience and cardiovascular protection. Bergamot Extract Cardiovascular and Metabolic Health Benefits Bergamot Extract and Metabolic Syndrome Bergamot extract is increasingly discussed in patients with: insulin resistance prediabetes obesity fatty liver disease metabolic syndrome Some studies suggest improvements in: fasting glucose insulin sensitivity inflammatory markers hepatic lipid metabolism This makes bergamot extract particularly attractive in broader longevity and metabolic optimization programs. Could Bergamot Extract Help Patients Who Cannot Tolerate Statins? Some individuals develop: muscle discomfort fatigue liver enzyme abnormalities statin intolerance In selected patients, physicians may consider bergamot extract as part of a broader integrative lipid management strategy. Some research suggests additive lipid-lowering effects when bergamot extract is combined with lower-dose statin therapy.^4^ Bergamot is the perfect agent to take if you cannot take statins, if you cannot tolerate statins, or if you simply prefer to take something instead of the statin. Patients should never stop prescription medications without physician guidance. Bergamot Extract Cardiovascular and Metabolic Health Benefits Infographic Typical Dosing Most clinical studies evaluating cholesterol support have used: 500–1,500 mg daily dosage of bergamot polyphenol extract 500 mg capsule, start with 1 per day, increase to twice daily after a week, then increase only if the cholesterol does not fall to desired level. Bergamot Extract 500 mg. Take 1 capsule twice daily. If needed, go to 1 capsule three times daily. Dosage guided by blood work results. Standardized extracts are generally preferred because potency varies considerably between products. Safety and Side Effects Bergamot extract is generally well tolerated. Potential side effects may include: gastrointestinal upset muscle cramps heartburn medication interactions Patients taking: statins anticoagulants diabetes medications antihypertensive medications should discuss supplementation with their physician before beginning therapy. Lifestyle Still Matters No supplement replaces: healthy nutrition exercise sleep optimization smoking cessation weight management metabolic control The greatest cardiovascular improvements occur when natural therapies are combined with comprehensive lifestyle modification and appropriate medical care. Bergamot extract should be viewed as one component of a broader preventive cardiovascular strategy. Related Topics Insulin Resistance: The Hidden Precursor to Cardiovascular Disease, Dementia, and Accelerated Aging Metabolic Syndrome: The Clinical Turning Point Uric Acid and Nitric Oxide Vitamin K and Cardiovascular Health Cacao and Heart Disease Selenium and Thyroid Function Bottom Line Bergamot extract is a promising natural supplement with growing evidence supporting its role in cholesterol management, metabolic health, and cardiovascular wellness. Its antioxidant and lipid-supportive properties may make it a valuable adjunct within physician-guided preventive medicine programs. As research continues to evolve, bergamot extract may become an increasingly important tool in integrative cardiovascular care. Become a Patient At Stages of Life Medical Institute, we provide comprehensive evaluations focused on metabolic health, cardiovascular prevention, hormone optimization, inflammation reduction, and longevity medicine. References Di Donna L, et al. Statin-like principles of bergamot fruit (Citrus bergamia): isolation of active polyphenols. J Nat Prod. 2009;72(7):1352-1354. PMID: 19507855. PubMed Reference 1 Mollace V, et al. Hypolipemic and hypoglycaemic activity of bergamot polyphenols. Fitoterapia. 2011;82(3):309-316. PMID: 21130811. PubMed Reference 2 Toth PP, et al. Bergamot and its role in cardiovascular prevention. Nutrients. 2020;12(11):3327. PMID: 33114285. PubMed Reference 3 Gliozzi M, et al. Bergamot polyphenolic fraction enhances rosuvastatin effects. Fitoterapia. 2013;90:69-77. PMID: 23856538. PubMed Reference 4 Navarra M, et al. Citrus bergamia polyphenols: biological activities and clinical potential. Planta Med. 2015;81(6):495-504. PMID: 25736451. PubMed Reference 5 Musolino V, et al. Bergamot polyphenols and vascular health. Pharmacol Res. 2020;159:104946. PMID: 32526249. PubMed Reference 6 Mollace R, et al. Bergamot polyphenolic fraction supplementation improves metabolic balance. Front Pharmacol. 2021;12:619436. PMID: 33716808. PubMed Reference 7 Leopoldini M, et al. Bergamot polyphenols and oxidative stress reduction. Food Chem Toxicol. 2010;48(6):1503-1510. PMID: 20381560. PubMed Reference 8 Carresi C, et al. Natural antioxidants and metabolic syndrome. Oxid Med Cell Longev. 2018;2018:8162518. PMID: 29765420. PubMed Reference 9 Mollace V, et al. Bergamot-derived polyphenolic fraction and lipid metabolism. Phytother Res. 2019;33(7):1805-1813. PMID: 31066113. PubMed Reference 10 REFERENCES The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Metabolic Syndrome: The Clinical Turning Point You Can Still Reverse
If insulin resistance is the silent beginning, metabolic syndrome is the moment it becomes clinically visible. This is where risk becomes measurable—and intervention becomes urgent. Metabolic syndrome significantly increases the likelihood of: Cardiovascular disease¹ Type 2 diabetes² Stroke¹ Cognitive decline³ Importantly, this stage remains highly reversible with appropriate care. What Is Metabolic Syndrome? Metabolic syndrome is diagnosed when three or more of the following are present⁴: Abdominal obesity (increased waist circumference) Elevated triglycerides Low HDL cholesterol Elevated blood pressure Elevated fasting glucose At its core, metabolic syndrome represents advanced insulin resistance expressed across multiple organ systems. Figure 1. Metabolic Syndrome Diagnostic Criteria Waist HDL Glucose Blood Pressure Why Metabolic Syndrome Matters 1. Cardiovascular Risk Accelerates Rapidly Metabolic syndrome promotes: Endothelial dysfunction¹ Atherosclerosis¹ Increased clotting risk¹ Patients often feel well while vascular disease progresses silently. 2. Progression to Diabetes Without intervention, many patients progress to type 2 diabetes². This transition reflects: Declining pancreatic reserve Persistent hyperglycemia Increasing metabolic injury 3. Brain and Cognitive Effects Metabolic syndrome is associated with: Reduced cerebral blood flow³ Increased dementia risk³ Impaired cognitive performance³ Figure 2. Progression from Insulin Resistance to Metabolic Syndrome What Drives Metabolic Syndrome? The same factors that initiate insulin resistance continue to push disease forward: Excess refined carbohydrates and sugar⁵ Sedentary lifestyle⁶ Visceral fat accumulation⁵ Chronic inflammation⁶ Hormonal imbalance⁶ How to Detect It Early Routine screening should include: Waist circumference Lipid panel (triglycerides, HDL) Blood pressure Fasting glucose However, earlier detection is possible with: Fasting insulin HOMA-IR Triglyceride/HDL ratio These allow identification of metabolic dysfunction before full syndrome develops. Figure 3. Clinical pathway for detecting and reversing metabolic syndrome. Early screening identifies risk factors before disease progression, allowing targeted intervention and potential reversal of metabolic dysfunction. Clinical Approach to Reversal 1. Nutrition Reduce processed carbohydrates and sugars⁵ Emphasize protein and healthy fats Stabilize blood glucose patterns 2. Exercise Resistance training improves insulin sensitivity⁶ Aerobic exercise reduces visceral fat⁶ 3. Weight Reduction Even modest weight loss improves metabolic markers significantly⁵. 4. Targeted Supplementation Evidence-supported options may include: Berberine Omega-3 fatty acids Magnesium Alpha-lipoic acid 5. Hormonal Optimization Addressing these can significantly improve metabolic outcomes⁶: Testosterone Thyroid function Cortisol Related Topics Insulin Resistance and Early Detection Uric Acid and Cardiovascular Risk Chronic Inflammation and Aging Hormonal Regulation and Metabolic Health Bottom Line Metabolic syndrome represents a critical, actionable turning point in chronic disease progression. At this stage, intervention is often highly effective in reversing risk and restoring metabolic health¹⁻². Call to Action At Stages of Life Medical Institute, we specialize in identifying and reversing metabolic syndrome before it progresses to diabetes or cardiovascular disease. 👉 Become a Patient:https://stagesoflifemedicalinstitute.com References Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008. https://pubmed.ncbi.nlm.nih.gov/18535151/ Alberti KG, et al. Harmonizing the metabolic syndrome. Circulation. 2009. https://pubmed.ncbi.nlm.nih.gov/19805654/ Yates KF, et al. Metabolic syndrome and cognitive decline. J Alzheimers Dis. 2012. https://pubmed.ncbi.nlm.nih.gov/22214744/ National Cholesterol Education Program (NCEP). ATP III guidelines. https://pubmed.ncbi.nlm.nih.gov/12485966/ Kahn SE, et al. Mechanisms linking obesity to insulin resistance. Nature. 2006. https://pubmed.ncbi.nlm.nih.gov/17167471/ Samuel VT, Shulman GI. The pathogenesis of insulin resistance. Cell. 2012. https://pubmed.ncbi.nlm.nih.gov/22385956/ The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Insulin Resistance: The Hidden Precursor to Cardiovascular Disease, Dementia, and Accelerated Aging
Insulin Resistance: Hidden Driver of Heart Disease & Dementia Insulin Resistance: Why It Matters More Than You Think Most patients associate insulin resistance with diabetes. Clinically, that is far too late. Insulin resistance is often present 10–20 years before diabetes develops¹, quietly driving: Atherosclerosis Hypertension Chronic inflammation Neurodegeneration Visceral fat accumulation By the time fasting glucose rises, significant metabolic injury has already occurred. Insulin resistance occurs when cells no longer respond effectively to insulin, forcing the pancreas to produce increasing amounts to maintain normal glucose levels. This results in: Chronically elevated insulin (hyperinsulinemia)⁴ Impaired glucose uptake⁷ Increased fat storage⁵ Endothelial dysfunction¹ Over time, this creates a systemic metabolic imbalance affecting nearly every organ system. Figure 1. Comparison of normal insulin signaling versus insulin resistance. In normal physiology, insulin binds to cellular receptors and facilitates glucose uptake into muscle and fat cells. In insulin resistance, cellular response is impaired, resulting in reduced glucose uptake, compensatory hyperinsulinemia, and increased metabolic stress. 1. Cardiovascular Disease Insulin resistance contributes directly to: Endothelial dysfunction¹ Increased triglycerides⁸ Reduced HDL⁸ Small dense LDL formation⁸ These changes accelerate plaque formation and vascular inflammation¹. 2. Dementia and Cognitive Decline Insulin plays a critical role in brain signaling. Insulin resistance in the brain is associated with: Reduced neuronal glucose uptake² Amyloid accumulation² Tau phosphorylation² This has led to the concept of Alzheimer’s disease as “Type 3 diabetes.”² 3. Accelerated Aging Chronic hyperinsulinemia promotes: Oxidative stress³ Mitochondrial dysfunction³ Chronic inflammation³ These processes accelerate biological aging³,¹⁰. Figure 2. Insulin resistance acts as a central driver of systemic disease. Chronic hyperinsulinemia contributes to endothelial dysfunction, cardiovascular disease, neurodegeneration, visceral fat accumulation, and chronic inflammation—linking metabolic dysfunction to accelerated aging. How Insulin Resistance Develops The most common contributors include: Excess refined carbohydrates and sugar⁵ Sedentary lifestyle⁷ Visceral adiposity⁵ Chronic stress (cortisol elevation)⁷ Poor sleep⁷ Importantly, insulin resistance can occur in normal-weight individuals⁹. How to Detect Insulin Resistance Early Standard glucose testing alone is insufficient. More sensitive markers include: Fasting insulin HOMA-IR Triglyceride/HDL ratio⁸ Hemoglobin A1c HOMA-IR Calculation HOMA-IR = Fasting Insulin × Fasting Glucose ÷ 405 Even modest elevations suggest early metabolic dysfunction⁶. Figure 3. Clinical pathway for early detection and intervention in insulin resistance. Patients often begin with nonspecific symptoms or normal standard labs. Early testing—such as fasting insulin, HOMA-IR, and lipid ratios—identifies metabolic dysfunction before progression to metabolic syndrome and diabetes, allowing timely intervention. Clinical Approach to Reversal 1. Nutrition Reduce refined carbohydrates and sugars⁵ Emphasize protein and healthy fats Consider time-restricted eating 2. Exercise Resistance training improves insulin sensitivity⁷ Aerobic exercise enhances glucose uptake⁷ 3. Weight and Visceral Fat Reduction Reducing abdominal fat is central to reversing insulin resistance⁵. 4. Targeted Supplementation Evidence-supported options may include: Berberine Magnesium Alpha-lipoic acid Omega-3 fatty acids These support insulin signaling and metabolic balance⁷. 5. Hormonal Optimization Where appropriate: Testosterone optimization Thyroid balance Cortisol regulation These can significantly influence insulin sensitivity⁷. Related Topics Metabolic Syndrome and Cardiovascular Risk Uric Acid and Endothelial Dysfunction Chronic Inflammation and Aging Hormonal Influence on Metabolic Health Bottom Line Insulin resistance is not simply a precursor to diabetes—it is a central driver of cardiovascular disease, dementia, and aging¹⁻³. Early detection using appropriate laboratory markers allows for timely, effective intervention, often reversing the process before irreversible damage occurs⁶. Call to Action At Stages of Life Medical Institute, we focus on early detection and reversal of insulin resistance through comprehensive metabolic testing and personalized care. 👉 Become a Patient:https://stagesoflifemedicalinstitute.com References Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. https://pubmed.ncbi.nlm.nih.gov/3056758/ de la Monte SM. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009;42(8):475–481. https://pubmed.ncbi.nlm.nih.gov/19754970/ Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133–2223. https://pubmed.ncbi.nlm.nih.gov/30067154/ Shanik MH, et al. Insulin resistance and hyperinsulinemia. Diabetes Care. 2008;31(Suppl 2):S262–S268. https://pubmed.ncbi.nlm.nih.gov/18227495/ Kahn SE, et al. Mechanisms linking obesity to insulin resistance. Nature. 2006;444(7121):840–846. https://pubmed.ncbi.nlm.nih.gov/17167471/ Craft S. Insulin resistance and cognitive decline. J Alzheimers Dis. 2005;7(1):53–62. https://pubmed.ncbi.nlm.nih.gov/15750214/ Samuel VT, Shulman GI. The pathogenesis of insulin resistance. Cell. 2012;148(5):852–871. https://pubmed.ncbi.nlm.nih.gov/22385956/ Grundy SM. Metabolic syndrome update. Circulation. 2008;117(25):e739–e743. https://pubmed.ncbi.nlm.nih.gov/18574054/ Lebovitz HE. Insulin resistance: definition and consequences. Exp Clin Endocrinol Diabetes. 2001;109(Suppl 2):S135–S148. https://pubmed.ncbi.nlm.nih.gov/11460565/ Ferrannini E, et al. Insulin resistance and aging. J Gerontol. 1993;48(Spec No):M43–M47. https://pubmed.ncbi.nlm.nih.gov/8409249/ The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Levator Scapulae Syndrome: The Culprit of Neck Pain
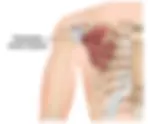
Levator scapulae syndrome is one of the most underdiagnosed causes of chronic neck and shoulder pain. A 2026 review in Frontiers in Public Health found that 55-69% of computer users now report neck pain, and clinical research shows up to 93% of office workers with neck and scapular complaints have measurable tightness in the levator scapulae muscle. Patients describe a deep ache at the base of the neck, pain that travels into the shoulder blade, and a tight "rope" running from the skull down to the shoulder. The good news: when correctly identified, levator scapulae syndrome is one of the more treatable musculoskeletal pain conditions. This guide covers what the levator scapulae actually does, why it gets injured, the symptoms to watch for, and the stretches and treatments that work — based on what we see every week at Stages of Life Medical Institute in Longwood, Florida. Understanding the Anatomy Tendinitis at this point causes pain in the shoulder, neck and may radiate to the pectoral muscles; referred pain to the thumb, index and middle fingers. The levator scapulae is a slender, strap-like muscle on the side and back of the neck. It originates from the transverse processes of the first four cervical vertebrae (C1-C4) and inserts on the upper inner border of the scapula. Its job, as the name suggests, is to elevate the shoulder blade — every time you shrug, look down at your phone, or carry a heavy bag, your levator scapulae fires. Because it spans the neck and shoulder simultaneously, it is uniquely vulnerable to overuse. A 2022 study in the International Journal of Occupational Safety and Ergonomics found that 90% of office workers with neck and scapular pain show measurable scapular dyskinesis — abnormal scapular movement that overloads the levator scapulae. The brachial plexus, the nerve bundle that supplies the arm, runs in close proximity, which is why levator scapulae problems can radiate pain or tingling into the shoulder, upper back, and even the fingers. The Levator Scapulae Muscle is important in posture but is delicate and easily injured. Understanding why the levator scapulae becomes injured is the first step toward effective treatment. In our clinic, most cases trace back to a small number of common triggers that build up over weeks or months of repetitive strain. Skeletal anatomy showing levator scapulae muscle location. What Causes Levator Scapulae Syndrome? Nerve entrapment at the levator scapulae can result from several factors, including: Forward Head Posture and Desk Work: Roughly 43% of office workers with neck pain show measurable forward head posture, and 100% show rounded shoulders (Tandfonline, 2022). Every inch the head shifts forward roughly doubles the load on the posterior neck muscles — the levator scapulae carries that load. Repetitive and Asymmetric Strain: Carrying a bag on the same shoulder, painting ceilings, sleeping on your stomach with the head turned, holding a phone between ear and shoulder, or working at a tilted or off-axis monitor all chronically shorten the levator scapulae. The body adapts to whatever position it spends the most time in, and the levator scapulae rarely wins that battle. Stress and Bracing: Mental stress drives unconscious shoulder elevation, and the levator scapulae is one of the first muscles to grip when we tense up. Over weeks of repetitive bracing, that elevated resting tone becomes the new baseline and the muscle stays tight even at rest. We see the strongest correlation in patients with high-demand jobs, poor sleep, or unresolved anxiety. Cervical or Shoulder Injury: Whiplash, slip-and-fall accidents, lifting injuries, and rotator-cuff problems all disturb scapular mechanics and overload the levator scapulae. Roughly half of whiplash patients still report neck pain months after the original event, frequently centered on the levator attachment at the upper inner border of the shoulder blade. Degenerative Cervical Changes: Arthritic changes at the C4-C5 and C5-C6 segments are common with age and can refer pain into the levator scapulae and trigger compensatory tightness around the muscle. We see this pattern most often in patients over 50, especially those with a history of prolonged desk work or repetitive overhead labor. Understanding these triggers is the first step toward effective treatment. The good news is that for most patients, levator scapulae syndrome responds well to conservative care — stretching, postural correction, and targeted manual therapy. Surgery is almost never indicated. The pain is often worse with rotation to the opposite side Symptoms to Watch For Identifying the symptoms of nerve entrapment in the levator scapulae area is essential for timely intervention. Common symptoms include: Localized Pain: Sharp or dull pain localizing in the neck and upper shoulder region. Radiating Pain: Discomfort that travels down the arm, potentially into the shoulder. Numbness or Tingling: A tingling sensation in the arm or hand, indicating nerve compression. Muscle Weakness: Difficulty raising the arm or gripping objects due to muscle weakness linked to nerve irritation. If symptoms persist beyond 2 weeks, radiate down the arm, or include numbness, tingling, or weakness, see a clinician. Persistent radicular symptoms can indicate cervical nerve root involvement and warrant imaging. Patients in the Orlando and Longwood area can request an evaluation through our Pain Management program at Stages of Life Medical Institute. Therapeutic massage combined with oral anti-inflammatories, muscle relaxants and nerve blocks are often used, in conjunction. This problem is benign, non-life or limb threatening, but it is truly, a pain in the neck. Diagnosis and Treatment Options To diagnose nerve entrapment, healthcare providers will conduct a comprehensive medical history and physical examination. Techniques such as MRI or ultrasound may be employed to evaluate the structures near the levator scapulae. Once diagnosed, effective treatment options include: Physical Therapy: A skilled physical therapist combines soft-tissue release, scapular stabilization (lower-trapezius and serratus-anterior strengthening), and motor re-education. A 2024 Cochrane review of conservative care for chronic neck pain found supervised exercise therapy reduces pain scores by 30 to 50% versus usual care, with effects holding at 12-month follow-up. Chiropractic Care: Targeted cervical and upper-thoracic adjustments can restore segmental motion and reduce the compensatory load on the levator scapulae. We see the best outcomes when chiropractic is paired with active rehab, not used as a standalone fix. Refer-in to a chiropractor with neuromusculoskeletal training rather than a pure-adjustment practice. Massage Therapy: Therapeutic massage can reduce muscle tension and promote relaxation in the affected area. Research shows that around 70% of patients experience relief from chronic neck pain following regular massage. Pain Management: Anti-inflammatory medications or corticosteroid injections may temporarily relieve pain and swelling. An estimated 30 to 90% of those suffering from neck pain find relief through these medications, and injections often provide immediate relief. Home Care: Utilizing heat or cold packs, along with gentle stretching exercises at home, may assist in symptom management. Topical Medications: Topical anti-inflammatories deliver medication straight to the levator scapulae insertion at the upper inner border of the scapula, producing concentrated relief without systemic side effects. The two we use and recommend daily in clinic are Kink-Ease MSM Salve (developed by Dr. Klein and stocked through our practice) and Voltaren (topical diclofenac, now available over the counter). Alternating the two through the day tends to outperform either alone. Voltaren topical diclofenac is now OTC available. Kink-Ease MSM Cream 16 oz. $44.95/bottle Three for $114.62 Recognizing the appropriate interventions is key to alleviating pain and restoring function. Preventative Measures Preventing nerve entrapment requires attention to posture and lifestyle habits. Here are some strategies to consider: Maintain Proper Posture: Ensure your sitting and standing positions support a neutral spine. Ergonomically designed office equipment can greatly enhance posture. Incorporate Regular Exercise: Engage in activities that promote flexibility and strength in the neck and shoulders, reducing tension and future pain episodes. Take Frequent Breaks: For those in desk jobs, taking regular breaks to stand, move around, and stretch can combat muscle tightness. A break every hour can significantly reduce discomfort. Manage Stress: Use relaxation techniques such as yoga, meditation, or deep breathing exercises to lessen muscle tension and anxiety. Implementing these preventative measures can significantly decrease the likelihood of encountering neck and shoulder pain in the future. Levator Scapulae Stretches You Can Do at Home These three stretches form the at-home protocol we recommend to patients. Do each one 2 to 3 times per day, hold each stretch for 30 seconds, and repeat both sides even if only one side hurts. Classic Levator Scapulae Stretch: Sit tall. Drop your right ear toward your right armpit. Rotate your chin 45 degrees down toward your right armpit, as if smelling under your arm. Place your right hand on top of your head and apply gentle downward pressure. Reach the left arm down and press the fingertips toward the floor to anchor the left scapula. Hold 30 seconds. You should feel a long, ropey stretch from the base of the skull to the inner shoulder blade. Repeat on the other side. Wall-Anchored Scapular Set: Stand with your back flat against a wall, heels 2 to 3 inches out. Press the back of your head, upper back, and tailbone into the wall. Slide your arms up the wall in a goalpost position, keeping the backs of the wrists touching the wall. Hold 5 seconds, lower, repeat 10 times. This trains the lower trapezius to take load off the levator scapulae. Self-Massage with a Lacrosse Ball: Place a lacrosse ball or tennis ball between your upper inner shoulder blade and a wall. Slowly roll the ball over the tender area for 60 to 90 seconds. Avoid pressing directly on the spine. Mild discomfort is normal; sharp pain is not. Frequently Asked Questions How long does levator scapulae syndrome take to heal? Most uncomplicated cases resolve in 2 to 6 weeks with consistent stretching, postural correction, and topical anti-inflammatories. Cases involving an underlying cervical issue or chronic ergonomic strain may take 8 to 12 weeks of structured rehabilitation. What is the ICD-10 code for levator scapulae syndrome? Levator scapulae syndrome typically codes under M25.819 (other specified disorder of unspecified joint, shoulder region) or M62.838 (other muscle spasm) depending on presentation. M54.2 (cervicalgia) is used when neck pain is the dominant symptom. Your clinician will choose the most accurate code based on exam findings. Can levator scapulae syndrome cause headaches? Yes, and this is one of the most missed diagnoses we see. The levator scapulae shares fascial and neural connections with the suboccipital muscles at the base of the skull. Chronic levator scapulae tension routinely refers into a tight band that wraps around the back of the head and behind the eyes, presenting as a tension-type headache. Is heat or ice better for levator scapulae pain? In the first 24 to 48 hours after a flare-up, ice for 15 minutes at a time helps reduce inflammation. After that, moist heat from a hot shower or a heated rice pack relaxes the muscle and is more therapeutic. The rule we tell patients: ice for swelling, heat for stiffness. Taking Action on Your Health Levator scapulae syndrome is one of the easier neck pain diagnoses to miss and one of the easier to fix once it is spotted. The pattern is recognizable: a tight rope from skull to shoulder blade, pain that worsens when you turn the head away from it, and a stiff neck most mornings. The protocol in this guide — postural correction, the three home stretches above, topical anti-inflammatories, and short courses of NSAIDs — resolves most cases in 2 to 6 weeks. If your pain has lasted longer than two weeks, radiates down the arm, or comes with numbness, tingling, or weakness, that is a clinic visit. We see and treat levator scapulae syndrome every week at Stages of Life Medical Institute in Longwood, Florida. Call 407-679-3337 or request a new patient appointment to be seen this week. REFERENCES: Henry C, Reidler J. Levator Scapulae Syndrome. StatPearls Publishing; 2024. https://www.ncbi.nlm.nih.gov/books/NBK556128/ Cohen SP, Hooten WM. Advances in the diagnosis and management of neck pain. BMJ. 2017;358:j3221. https://www.bmj.com/content/358/bmj.j3221 Khaledi A, et al. Investigation of postural abnormalities in office workers with neck pain. International Journal of Occupational Safety and Ergonomics. 2022;28(4). https://www.tandfonline.com/doi/full/10.1080/10803548.2021.1916206 Physiopedia. (n.d.). Levator scapulae syndrome. https://www.physio-pedia.com/Levator_Scapulae_Syndrome OrthoFixar. (n.d.). Overview of levator scapulae pain causes and treatment. https://orthofixar.com/for-patient/levator-scapulae-pain-treatment Release Muscle Therapy. (n.d.). Levator scapulae pain: Unraveling the mystery and finding relief. https://releasemuscletherapy.com/pain-in-levator-scapulae-relief Leagrave Therapy. (n.d.). Neck pain and levator scapulae syndrome. https://www.leagravetherapy.co.uk/levator-scapulae-syndrome-and-neck-pain Physiopedia. (n.d.). Levator scapulae. https://www.physio-pedia.com/Levator_Scapulae Physio.co.uk. (n.d.). Levator scapulae syndrome. https://www.physio.co.uk/what-we-treat/musculoskeletal/conditions/shoulder/levator-scapulae-syndrome.php Bel Marra Health. (n.d.). How is levator scapulae pain related to stiff neck and how to relieve the pain?. https://www.belmarrahealth.com/levator-scapulae-pain-causes-symptoms-treatment-exercises Physical Therapy. (n.d.). Levator scapulae muscle pain: Cause, treatment, exercise. https://physical-therapy.us/levator-scapulae-muscle-pain Physio Insights. (n.d.). Levator scapulae pain. https://physioinsights.com/2020/12/31/levator-scapulae-pain NSMI. (n.d.). Levator scapulae syndrome - Muscular injuries. https://www.nsmi.org.uk/articles/muscular-injuries/levator-scapulae-syndrome.html Subscribe to our Blog Dr Klein's Facebook Page https://www.facebook.com/stagesoflifemedicalinstitute David S. Klein, MD FACA FACPM David S. Klein, MD, FACA, FACPM 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com David S. Klein, MD Functional Medicine Physician
- Hormone Replacement Therapy (HRT) and Osteoporosis: Prevention and Treatment
Introduction Osteoporosis is not simply a disease of aging—it is largely a disease of hormonal decline. The reduction in estrogen (and testosterone) directly accelerates bone loss, increasing fracture risk, loss of independence, and mortality.¹ While calcium and vitamin D are important, they are supportive—not primary—drivers of bone health. Hormonal signaling remains central. This is where Hormone Replacement Therapy (HRT) plays a critical role in both prevention and treatment of osteoporosis. Bone Mass, Bone Density Decrease at Menopause Bone remodeling is a balance between: Osteoclasts (bone breakdown) Osteoblasts (bone formation) Estrogen suppresses osteoclast activity and inflammatory signaling, while testosterone supports bone formation and converts to estrogen via aromatization.² ³ Clinical takeaway: Hormone decline shifts this balance toward bone loss. Why Hormone Decline Leads to Osteoporosis The most rapid bone loss occurs: Within 5–10 years after menopause Gradually in men with declining testosterone Mechanisms include: Increased osteoclast activity Reduced osteoblast function Loss of bone strength and architecture This explains why calcium alone does not prevent osteoporosis.⁴ HRT for Prevention of Osteoporosis Estrogen Therapy (Women) Estrogen therapy: Prevents bone loss Maintains bone density Reduces fracture risk by 30–50%⁵ It is most effective when initiated near menopause.⁶ Testosterone Therapy (Men and Women) Testosterone: Supports bone formation Improves muscle mass (reducing fall risk) Converts to estrogen (critical for bone health)³ Low testosterone is strongly associated with osteoporosis and fracture risk.⁷ HRT for Treatment of Osteoporosis HRT can: Slow or halt bone loss Improve bone mineral density Enhance bone quality Unlike antiresorptive drugs, HRT restores physiologic hormonal signaling.⁸ HRT vs Conventional Osteoporosis Treatments Therapy Mechanism Consideration Bisphosphonates Reduce bone breakdown Long-term remodeling concerns Denosumab Inhibits RANKL Rebound bone loss if stopped SERMs Partial estrogen effect Limited scope HRT Restores hormones Requires individualized care HRT addresses the underlying hormonal deficiency, not just bone turnover.⁶ Safety Considerations Concerns about HRT largely stem from early interpretations of the Women's Health Initiative.⁹ Current understanding: Risk varies by timing, formulation, and patient selection Transdermal estrogen has a more favorable safety profile Appropriate patient selection makes HRT a safe and effective option for many individuals.¹⁰ Comprehensive Bone Health Strategy Best outcomes occur when HRT is combined with: Vitamin D optimization Calcium intake Magnesium and vitamin K2 Resistance training Fall prevention Clinical Pathway: When to Consider HRT Consider evaluation in patients with: Osteopenia or osteoporosis Early menopause Hormonal symptoms History of fracture Bottom Line Osteoporosis is fundamentally a hormone-driven condition. HRT restores the physiologic signals that regulate bone remodeling, making it one of the most effective strategies for preventing and treating osteoporosis when appropriately prescribed. Become a Patient Comprehensive hormone and bone health evaluation is available through Stages of Life Medical Institute. 🔗 https://www.stagesoflifemedicalinstitute.com/ References Cauley JA. Estrogen and bone health in men and women. JAMA. 2015;313(13):1336–1345. Riggs BL, Khosla S, Melton LJ. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. 2002;23(3):279–302. Khosla S. Pathogenesis of age-related bone loss in humans. J Gerontol A Biol Sci Med Sci. 2013;68(10):1226–1235. Heaney RP. Calcium, dairy products and osteoporosis. J Am Coll Nutr. 2000;19(2 Suppl):83S–99S. Rossouw JE, et al. Risks and benefits of estrogen plus progestin. JAMA. 2002;288(3):321–333. The North American Menopause Society. The 2022 hormone therapy position statement. Menopause. 2022;29(7):767–794. Snyder PJ, et al. Effects of testosterone treatment in older men. N Engl J Med. 2016;374(7):611–624. Compston JE, et al. Osteoporosis. Lancet. 2019;393(10169):364–376. Anderson GL, et al. Effects of estrogen plus progestin in postmenopausal women. JAMA. 2004;291(14):1701–1712. Canonico M, et al. Postmenopausal hormone therapy and cardiovascular risk. Circulation. 2007;115(7):840–845. The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Strontium and Osteoporosis: A Natural Element with Clinical Potential
Strontium for Osteoporosis: Natural Bone Strength Support Introduction Osteoporosis is one of the most common and silent conditions affecting aging adults. Bone loss often progresses without symptoms until a fracture occurs. Normal vs Osteoporotic Bone Structure Comparison While conventional medications are widely prescribed, many patients seek more natural approaches to preserving bone density. One element gaining attention is strontium—a naturally occurring mineral found in soil, groundwater, and trace amounts in food¹. Its appeal lies in a simple premise: a naturally derived substance that may support bone strength through physiologic mechanisms. What Is Strontium? Strontium is a trace mineral chemically similar to calcium². It is naturally present in: Groundwater Soil and plant-based foods Small amounts within human bone Because of its similarity to calcium, strontium can be incorporated into bone tissue, where it influences the balance between bone formation and breakdown². How Strontium Supports Bone Health Unlike many treatments that act in a single direction, strontium appears to work through a dual mechanism: Stimulates osteoblasts → increases bone formation³ Inhibits osteoclasts → reduces bone breakdown³ This balanced effect makes it particularly interesting in the management of osteoporosis. Clinical Evidence and Real-World Use The most studied form is strontium ranelate, which has been evaluated in large randomized controlled trials. These studies demonstrated: Increased bone mineral density Reduced vertebral and non-vertebral fracture risk⁴⁻⁶ However: The Strontium Renelate product is not FDA-approved in the United States Use has been restricted due to cardiovascular risk concerns⁷ Strontium Citrate is readily available, however, and is very inexpensive. Strontium citrate is generally well tolerated in healthy individuals when used at typical supplemental doses (commonly ~680 mg elemental strontium daily). Unlike prescription Strontium ranelate, it has not been subjected to large randomized safety trials—so much of its safety profile is extrapolated rather than definitively established. In clinical practice, many patients use strontium citrate, an over-the-counter form. While widely used, it does not have the same level of robust clinical trial evidence⁸. A Critical Point: Bone Density Measurements Strontium has an important and often overlooked effect: ➡️ It can modestly artificially increase bone density readings on DEXA scans⁹ This occurs because strontium alters X-ray attenuation due to its higher atomic weight compared to calcium. Clinical implications: Bone density may appear improved True bone strength may be overestimated Interpretation of DEXA results in patients using strontium requires careful clinical correlation. Is Strontium Deficiency the Cause of Osteoporosis? There is currently no definitive evidence that osteoporosis is caused by strontium deficiency. There are no standardized lab tests for deficiency It is not considered an essential nutrient in the traditional sense¹⁰ However, environmental exposure varies, and strontium may still play a supportive role in bone physiology. Strontium Citrate 300 mg elemental per capsule Safety and Practical Use Strontium is generally well tolerated when used appropriately, but important precautions apply. Use cautiously in: Cardiovascular disease, severe History of thrombosis Significant renal impairment⁷ Practical considerations: Take separately from calcium Common clinical dosing: ~680 mg daily (strontium citrate) Strontium Citrate: Common Side Effects (Usually Mild) Reported adverse effects are typically infrequent and mild: Gastrointestinal discomfort (nausea, loose stools) Headache Occasional skin rash These are not dose-limiting in most patients and often improve with dose adjustment. Where Strontium Fits in Treatment Strontium may be appropriate in: Patients seeking non-pharmaceutical approaches Those intolerant of conventional therapies As part of a comprehensive bone health strategy Optimal bone care should include: Vitamin D-3 optimization Adequate calcium intake, calcium hydroxyapatite Vitamin K-2 intake Hormonal evaluation when indicated Weight-bearing exercise Hormone Replacement Therapy, if levels are low Bottom Line Strontium is a naturally occurring element with measurable effects on bone metabolism. While not a first-line therapy, it represents a valuable adjunct in selected patients when used with proper clinical oversight. Become a Patient If you are concerned about bone loss or osteoporosis, we provide comprehensive evaluation and individualized treatment plans. 👉 https://www.stagesoflifemedicalinstitute.com References (PubMed Indexed) Trace elements in human nutrition – Nielsen FH. Ultratrace elements in nutrition. Annu Rev Nutr. 1984. PMID: 6085717 Strontium incorporation into bone – Dahl SG et al. Incorporation and distribution of strontium in bone. Bone. 2001. PMID: 11502467 Bone remodeling physiology – Marie PJ. Strontium ranelate: a dual mode of action. Bone. 2005. PMID: 16046172 SOTI trial – Meunier PJ et al. Strontium ranelate reduces vertebral fractures. NEJM. 2004. PMID: 15356304 TROPOS trial – Reginster JY et al. Strontium ranelate reduces nonvertebral fractures. J Clin Endocrinol Metab. 2005. PMID: 16030160 Long term osteoporosis therapy outcomes – Reginster JY et al. Long-term effects of strontium ranelate. Osteoporos Int. 2008. PMID: 18266020 Cardiovascular risk with strontium ranelate – EMA safety review of strontium ranelate. Drug Saf. 2014. PMID: 24752455 Strontium citrate supplementation – Shorr RI et al. Dietary supplements and bone health. Am J Med. 2010. PMID: 20362756 DEXA scan artifact strontium – Blake GM et al. Effect of bone strontium on BMD measurements. J Clin Densitom. 2007. PMID: 17543560 Essential vs non essential trace elements – Mertz W. The essential trace elements. Science. 1981. PMID: 7022654 The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- What Is Sepsis? Recognizing the Symptoms and Early Signs of a Life-Threatening Infection
Introduction Sepsis is one of the most serious medical conditions we encounter—and one of the most misunderstood. It does not begin as a disease on its own, but rather as the body’s overwhelming and dysregulated response to an infection. Left untreated, it can progress rapidly, leading to organ failure and death. Understanding how sepsis develops, how it presents, and when to act can quite literally save a life. What Is Sepsis? Sepsis occurs when the body’s response to infection becomes excessive and begins to damage its own tissues and organs. Normally, the immune system fights infection in a controlled way. In sepsis, that response becomes amplified and unbalanced. Instead of protecting you, the immune system triggers widespread inflammation, blood vessel dysfunction, and impaired oxygen delivery. See Figure 1: Progression from Infection to Sepsis Sepsis Progression: From Infection to Septic Shock Explained How Does Sepsis Develop? Sepsis typically begins with a localized infection, such as: Urinary tract infections (UTIs) Pneumonia Skin infections Abdominal infections In older adults especially, a urinary tract infection may not always present with classic burning or urgency, which is why early recognition is so important. You can read more in our related article on [UTI diagnosis and PCR testing]. In clinical practice, urinary infections are a particularly important source—especially in older adults. As discussed in our related article on UTI diagnosis and PCR testing, infections may not always present with classic symptoms and can progress silently. When a urinary infection spreads beyond the bladder and begins affecting the whole body, it may progress into a dangerous condition known as [urosepsis]. Once bacteria—or sometimes viruses or fungi—enter the bloodstream, the body mounts an aggressive immune response. This leads to: Widespread inflammation Leaky blood vessels Reduced blood flow to vital organs The result is a cascade that can progress from infection → sepsis → severe sepsis → septic shock. How Does Sepsis Present? (Early Warning Signs) Sepsis can be difficult to recognize early because symptoms may appear subtle at first. Common Early Symptoms Fever or abnormally low temperature Rapid heart rate Rapid breathing Fatigue or weakness Confusion or altered mental status Progressive Symptoms Low blood pressure Decreased urine output Shortness of breath Skin discoloration or mottling In older patients, confusion or sudden weakness may be the first and only sign. This is one reason it is so important not to dismiss a subtle symptom as harmless, particularly when the underlying cause may be much more serious than it first appears. Our article on [causal vs. coincidental findings in medicine] explores this principle in more detail. This highlights an important principle discussed in our article on causal vs. coincidental findings: symptoms that appear mild may reflect a much more serious underlying process. See Figure 2: Early vs Late Symptoms of Sepsis Early vs Late Symptoms of Sepsis: Recognizing Warning Signs Who Is at Higher Risk? Certain populations are more vulnerable to developing sepsis: Adults over age 65 Individuals with diabetes or insulin resistance Patients with chronic kidney disease Those on immunosuppressive medications Individuals with indwelling catheters or recent procedures Patients with impaired metabolic health often have a more difficult time mounting a balanced immune response to infection. This is discussed further in our article on [insulin resistance and cardiovascular disease], which also explains how chronic metabolic dysfunction can affect recovery and resilience. Metabolic health plays a significant role. Conditions such as insulin resistance impair immune function and increase susceptibility to severe infection. How Is Sepsis Diagnosed? Sepsis is a clinical diagnosis supported by laboratory findings. Key Diagnostic Elements Evidence of infection Signs of organ dysfunction Common Laboratory Tests Complete blood count (CBC) Blood cultures Lactate levels (marker of tissue hypoxia) Kidney and liver function tests Urinalysis and culture In many cases, urinalysis followed by culture and sensitivity—with reflex to PCR when needed—can improve diagnostic accuracy, particularly in complex or recurrent infections. As emphasized in our UTI-focused discussions, PCR testing is particularly useful when traditional cultures are inconclusive or symptoms persist. How Is Sepsis Treated? Sepsis is a medical emergency. Early treatment dramatically improves survival. Core Treatment Principles 1. Rapid Antibiotic Therapy Broad-spectrum antibiotics are started immediately, then tailored once cultures return. 2. Intravenous Fluids Fluids help maintain blood pressure and improve circulation. 3. Oxygen and Organ Support Patients may require: Supplemental oxygen Mechanical ventilation Medications to support blood pressure (vasopressors) 4. Source Control The underlying infection must be addressed: Drainage of abscess Removal of infected devices Targeted antimicrobial therapy See Figure 3: Sepsis Treatment Pathway Figure 3. Sepsis treatment pathway illustrating the four critical pillars of care: rapid antibiotic therapy, intravenous fluids, organ and oxygen support, and source control. Early, coordinated intervention is essential to prevent progression to septic shock and organ failure. Why Early Recognition Matters Time is critical in sepsis. Each hour of delay in treatment increases mortality Early intervention can reverse the process Late-stage sepsis (septic shock) carries a significantly higher risk of death This reinforces a key clinical concept: recognizing patterns early—even when symptoms seem mild—can prevent catastrophic outcomes. Prevention: What Can Patients Do? While not all cases are preventable, risk can be reduced by: Prompt treatment of infections Good hygiene and wound care Managing chronic conditions (diabetes, kidney disease) Staying up to date on vaccinations Seeking care early when symptoms change unexpectedly In many patients, prevention begins with paying attention to early laboratory findings and responding before a localized infection becomes a systemic emergency. Our related article on [the risks of not treating bacteria in the urine] helps explain when observation may be reasonable and when delay may be dangerous. Related Topics Urinary Tract Infections and PCR Diagnosis Causal vs. Coincidental Findings in Clinical Medicine Insulin Resistance and Immune Dysfunction Urosepsis: When a UTI Becomes Life-Threatening Bottom Line Sepsis is not simply an infection—it is the body’s overwhelming and damaging response to one. It can begin subtly but progress rapidly, making early recognition and treatment essential. Understanding the warning signs, risk factors, and importance of timely care can make the difference between recovery and serious complications. Call to Action If you or a loved one are experiencing symptoms of infection that seem to be worsening—especially confusion, weakness, or rapid breathing—do not wait. At Stages of Life Medical Institute, we emphasize early diagnosis, precise laboratory evaluation, and individualized care to identify and treat infections before they progress to serious conditions like sepsis. 👉 Become a Patient: https://stagesoflifemedicalinstitute.com References Singer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. https://pubmed.ncbi.nlm.nih.gov/26903338/ Seymour CW, et al. Assessment of Clinical Criteria for Sepsis. JAMA. 2016;315(8):762–774. https://pubmed.ncbi.nlm.nih.gov/26903335/ Rhodes A, et al. Surviving Sepsis Campaign Guidelines. Intensive Care Med. 2017;43(3):304–377. https://pubmed.ncbi.nlm.nih.gov/28101605/ Rudd KE, et al. Global burden of sepsis. Lancet. 2020;395(10219):200–211. https://pubmed.ncbi.nlm.nih.gov/31954465/ Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. https://pubmed.ncbi.nlm.nih.gov/23984731/ Cecconi M, et al. Sepsis and septic shock. Lancet. 2018;392(10141):75–87. https://pubmed.ncbi.nlm.nih.gov/29937192/ Kumar A, et al. Duration of hypotension before treatment and survival in septic shock. Crit Care Med. 2006;34(6):1589–1596. https://pubmed.ncbi.nlm.nih.gov/16625125/ Kalil AC, et al. Management of adults with sepsis. Clin Infect Dis. 2016;63(5):e61–e111. https://pubmed.ncbi.nlm.nih.gov/27418577/ Hotchkiss RS, et al. Immunosuppression in sepsis. Nat Rev Immunol. 2013;13(12):862–874. https://pubmed.ncbi.nlm.nih.gov/24232462/ Evans L, et al. Surviving Sepsis Campaign 2021 update. Intensive Care Med. 2021;47:1181–1247. https://pubmed.ncbi.nlm.nih.gov/34605781/ The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Sepsis and the Urinary Tract: Why Diagnosis Fails—And How We Fix It
A Clinical Perspective on Geriatric UTI, Dipstick Limitations, and PCR-Based Detection Introduction Sepsis remains one of the most consequential—and frequently misrecognized—conditions in clinical medicine. It is not merely infection; it is a failure of physiologic regulation in response to infection, resulting in organ dysfunction and, if untreated, death.¹ Among all infectious sources, the urinary tract is one of the most common origins of sepsis, particularly in older adults. Yet paradoxically, it is also one of the most poorly diagnosed, especially in nursing homes and outpatient geriatric care.² The central issue is not a lack of testing—it is reliance on inadequate testing methods. What Sepsis Is (Clinically Speaking) Sepsis is best understood as: Infection + organ dysfunction The modern definition (Sepsis-3) describes sepsis as a dysregulated host response to infection leading to life-threatening organ dysfunction.¹ Once infection begins to impair cognition, renal function, respiration, circulation, or metabolic stability, the patient has crossed into sepsis. In older adults, this transition is often subtle, rapid, and easily missed. Clinical Significance Sepsis is both common and dangerous: Leading cause of hospitalization and mortality Disproportionately affects adults over age 65 Frequently underrecognized early Outcomes strongly tied to speed of diagnosis and treatment³ Early recognition is not simply beneficial—it is determinative. Why the Urinary Tract Matters Urinary infections are: Common in both community and institutional settings A frequent source of bacteremia and sepsis Often misdiagnosed in both directions This creates a dual clinical failure: Overdiagnosis → unnecessary antibiotics Underdiagnosis → progression to sepsis Geriatric UTI: The Diagnostic Paradox Older adults, particularly in nursing homes, present unique challenges: High prevalence of asymptomatic bacteriuria Atypical presentations (confusion, weakness, falls) Reduced reporting of urinary symptoms Importantly: Bacteriuria alone does not equal infection⁴ Treating colonization exposes patients to harm without benefit. The Nursing Home Problem In long-term care settings, a familiar pattern emerges: Patient develops confusion or decline Urine is tested Bacteria are found Antibiotics are prescribed This sequence is frequently incorrect. Cloudy urine, odor, or a positive dipstick are not sufficient criteria for diagnosing UTI.⁵ At the same time, true infection may be missed when symptoms are subtle. Where Traditional Testing Fails Urine Dipstick Limitations Dipsticks evaluate: Nitrites Leukocyte esterase They are screening—not diagnostic—tools. False negatives occur due to: Non–nitrite-producing organisms Frequent voiding Vitamin C interference Early infection Dilute urine⁶ False positives occur due to: Chronic inflammation Colonization Noninfectious causes of pyuria A negative dipstick does not exclude infection. A positive dipstick does not confirm infection. (See Figure 1: Limitations of Urine Dipstick Testing) Figure 1. Limitations of urine dipstick testing in UTI diagnosis, highlighting common causes of false negatives and false positives, particularly in geriatric patients. The Diagnostic Gap in UTI and Urosepsis Traditional urine culture: Requires 24–72 hours Depends on organism growth conditions May miss polymicrobial or fastidious organisms This creates a dangerous clinical window where: The patient deteriorates Diagnosis remains uncertain Treatment may be delayed or inappropriate A Modern Diagnostic Strategy: UA → Culture → Reflex PCR A practical and clinically sound approach is to begin with a routine urinalysis (UA) followed by culture and sensitivity (C&S), using these as the initial screening and organism-directed framework, and then reflexing to PCR-based testing when results are incongruent with the clinical picture. UA provides rapid insight into inflammatory activity (pyuria, hematuria, nitrites), while culture allows for organism identification and antibiotic susceptibility—still essential for targeted therapy. However, both are limited by sensitivity, growth requirements, and time delay. When a patient remains symptomatic despite a negative U, when cultures are repeatedly negative or slow-growing, or when the presentation is atypical—as is common in geriatric populations—PCR serves as a valuable second-line diagnostic tool, detecting microbial DNA independent of growth conditions and improving identification of polymicrobial or fastidious organisms.⁶–⁸ In this tiered model, UA and culture establish a cost-effective, standardized baseline, while PCR is deployed selectively to resolve diagnostic uncertainty and reduce both missed infections and inappropriate treatment.⁶–⁹ (See Figure 2: Diagnostic Pathway—Traditional vs PCR-Augmented Approach) Figure 2. A modern diagnostic approach to urinary tract infection, beginning with urinalysis, followed by culture and sensitivity, with reflex to PCR when findings are incongruent with clinical presentation. UTI Progression to Sepsis In older adults, progression may be subtle: Mild or absent urinary symptoms Functional decline Confusion or delirium Dehydration Renal dysfunction Hypotension Sepsis Early diagnostic ambiguity is common—and dangerous. (See Figure 3: Clinical Progression from UTI to Urosepsis) Clinical Decision Framework Step 1: Assess for symptoms Dysuria, urgency, frequency Fever, flank pain Hemodynamic instability Step 2: Recognize atypical presentations Confusion Weakness Functional decline Evaluate for systemic illness. Step 3: Apply appropriate testing Clinical Scenario Recommended Approach Classic UTI UA + culture Atypical geriatric UA + culture ± PCR Recurrent or refractory PCR strongly indicated Suspected sepsis Full evaluation + targeted diagnostics Prognosis Outcomes depend on: Speed of recognition Accuracy of diagnosis Timeliness of treatment Older adults face: Higher mortality Increased functional decline Greater recurrence risk³ However, early and accurate diagnosis significantly improves outcomes. Bottom Line Sepsis is a medical emergency defined by infection causing organ dysfunction. In older adults, urinary infections are a common pathway—but diagnosis is complicated by: Asymptomatic bacteriuria Atypical presentation Dipstick limitations Delayed or incomplete culture data A negative dipstick does not rule out infection. A positive dipstick does not confirm it. A modern, tiered diagnostic approach—UA followed by culture, with reflex to PCR when needed—offers a more accurate, clinically effective strategy. Call to Action If you or a loved one is experiencing: Recurrent urinary symptoms Confusion without clear explanation Persistent symptoms despite negative testing Concern for early infection or sepsis A more advanced diagnostic approach may be warranted. At Stages of Life Medical Institute, we integrate traditional clinical evaluation with advanced diagnostic tools—including PCR-based testing—to improve accuracy and patient outcomes. 👉 Become a Patient: https://stagesoflifemedicalinstitute.com References Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. https://pubmed.ncbi.nlm.nih.gov/26903338/ Hooton TM. Clinical practice. Uncomplicated urinary tract infection. N Engl J Med. 2012;366(11):1028–1037. https://pubmed.ncbi.nlm.nih.gov/22417256/ Rhee C, Dantes R, Epstein L, et al. Incidence and trends of sepsis in US hospitals. JAMA. 2017;318(13):1241–1249. https://pubmed.ncbi.nlm.nih.gov/28903154/ Nicolle LE, Gupta K, Bradley SF, et al. Clinical practice guideline for asymptomatic bacteriuria. Clin Infect Dis. 2019;68(10):e83–e110. https://pubmed.ncbi.nlm.nih.gov/30895288/ Loeb M, Bentley DW, Bradley S, et al. Development of minimum criteria for antibiotics in long-term care. Infect Control Hosp Epidemiol. 2001;22(2):120–124. https://pubmed.ncbi.nlm.nih.gov/11232875/ Simerville JA, Maxted WC, Pahira JJ. Urinalysis: A comprehensive review. Am Fam Physician. 2005;71(6):1153–1162. https://pubmed.ncbi.nlm.nih.gov/15791892/ Price TK, Dune T, Hilt EE, et al. Enhanced urine culture techniques. J Clin Microbiol. 2016;54(5):1216–1222. https://pubmed.ncbi.nlm.nih.gov/26962083/ Khasriya R, Sathiananthamoorthy S, Ismail S, et al. Bacterial colonization in LUTS. J Clin Microbiol. 2013;51(7):2054–2062. https://pubmed.ncbi.nlm.nih.gov/23637317/ Wolfe AJ, Brubaker L. “Sterile urine” reconsidered. Clin Microbiol Rev. 2015;28(3):719–733. https://pubmed.ncbi.nlm.nih.gov/25985967/ The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Avoid Vitamin D2, use Vitamin D3
And Why Vitamin D₃ Is Almost Always Preferred in Clinical Practice Many patients are surprised to learn that not all vitamin D supplements are the same. Labels may simply say “vitamin D,” but there are two very different forms commonly used: vitamin D₂ (ergocalciferol) and vitamin D₃ (cholecalciferol). From a physician’s standpoint, this distinction matters. Vitamin D₃ is almost always the superior choice for correcting deficiency and supporting long-term health.¹⁻³ Vitamin D₂ vs Vitamin D₃: A Brief Overview Vitamin D exists in two major supplemental forms: Vitamin D₂ (ergocalciferol) – derived from plant sources and fungi Vitamin D₃ (cholecalciferol) – biologically identical to the form synthesized in human skin following UVB exposure¹⁻³ Although both forms can raise vitamin D levels on laboratory testing, they are not biologically equivalent.²⁻⁶ The Biological Reality: D₂ and D₃ Are Not Interchangeable After ingestion, both vitamin D₂ and D₃ undergo hepatic and renal hydroxylation to become hormonally active. However, substantial differences exist in their pharmacokinetics and biologic behavior. Compared with vitamin D₂, vitamin D₃: Raises serum 25-hydroxyvitamin D more efficiently²⁻⁷ Has a longer circulating half-life⁶⁻⁹ Binds more tightly to vitamin D–binding protein¹⁰ Produces fewer inactive metabolites⁴⁻⁶ These differences translate into more predictable and durable clinical responses with vitamin D₃.³⁻⁷ A Critical and Often Overlooked Issue: Vitamin D₂ Can Suppress Vitamin D₃ Beyond being less effective, vitamin D₂ introduces an additional problem rarely discussed in patient-facing literature: it can actively reduce circulating vitamin D₃ levels. Controlled studies demonstrate that supplementation with vitamin D₂ increases serum 25-hydroxyvitamin D₂ while simultaneously decreasing endogenous 25-hydroxyvitamin D₃.²⁻³ This competitive effect is believed to result from shared hepatic hydroxylation pathways and weaker binding of D₂ metabolites to vitamin D–binding protein, leading to accelerated clearance of vitamin D₃, the body’s preferred and more biologically active form.²,³,¹⁰ Clinically, this means vitamin D₂ may paradoxically blunt overall vitamin D hormonal effectiveness, particularly in patients who require stable levels for bone, immune, neuromuscular, or cognitive health.²⁻⁴ Stability and Potency: Why D₂ Falls Short Vitamin D₂ is chemically less stable than vitamin D₃, which affects shelf stability, serum persistence, and dosing reliability.⁴⁻⁶ In contrast, vitamin D₃ produces: Higher peak serum 25-hydroxyvitamin D levels⁶⁻⁸ More sustained concentrations over time⁶⁻⁹ Greater consistency across repeated dosing⁷⁻⁹ These properties make vitamin D₃ far easier to monitor and titrate in clinical practice. What Physicians Observe in Practice Patients using vitamin D₂ frequently demonstrate: Smaller increases in serum 25-hydroxyvitamin D²⁻⁶ Faster declines after supplementation stops⁶⁻⁹ Greater inter-patient variability⁷⁻⁹ Vitamin D₃, by contrast, shows reliable repletion, predictable dose-response relationships, and superior long-term stability.³⁻⁹ Clinical Outcomes Favor Vitamin D₃ When clinically meaningful endpoints are examined—rather than short-term lab changes—vitamin D₃ consistently outperforms vitamin D₂. Vitamin D₃ has demonstrated superior effects on: Bone mineral density and fracture prevention¹¹⁻¹³ Parathyroid hormone suppression¹,¹¹ Muscle strength and fall reduction¹¹,¹⁴ Overall mortality risk in observational studies¹⁴ Vitamin D₂ has not shown equivalent consistency across these outcomes.⁴⁻⁶ Soy Free Vitamin D3 in Olive Oil with Vitamin K2 Why Vitamin D₂ Is Still Used Vitamin D₂ remains in use largely for historical and regulatory reasons: Earlier pharmaceutical formulations favored ergocalciferol It was easier to synthesize commercially decades ago Some prescription products still contain high-dose D₂ and doctors often 'disgorge bad habits, slowly.' Soy Free Vitamin D3 in Olive Oil However, availability does not imply equivalence, and modern evidence overwhelmingly favors vitamin D₃.³⁻⁷ Safety, Dosing, and Monitoring Both forms are fat-soluble and require clinical oversight at higher doses. However, because vitamin D₃ mirrors endogenous hormone physiology, its dose-response behavior is better understood and more predictable.¹,³,¹² This allows for safer targeting of optimal blood levels while minimizing toxicity risk.¹,¹² The Takeaway Although vitamin D₂ and vitamin D₃ share a name, they are not interchangeable therapies. From a physician’s perspective: Vitamin D₃ is more potent More stable Longer lasting More physiologically appropriate Most importantly, vitamin D₂ may interfere with vitamin D₃ itself, undermining effective hormonal signaling.²⁻⁴ For patients seeking durable improvements in bone health, immune resilience, muscle performance, and long-term disease prevention, vitamin D₃ is almost always the correct clinical choice. References Holick MF. Vitamin D deficiency. N Engl J Med. 2007.https://pubmed.ncbi.nlm.nih.gov/17634462/ Armas LAG, Hollis BW, Heaney RP. Vitamin D₂ is much less effective than vitamin D₃ in humans. J Clin Endocrinol Metab. 2004.https://pubmed.ncbi.nlm.nih.gov/15240628/ Tripkovic L, et al. Comparison of vitamin D₂ and vitamin D₃ supplementation. Am J Clin Nutr. 2012.https://pubmed.ncbi.nlm.nih.gov/22552031/ Houghton LA, Vieth R. The case against ergocalciferol (vitamin D₂). Am J Clin Nutr. 2006.https://pubmed.ncbi.nlm.nih.gov/16632698/ Heaney RP, et al. Vitamin D₃ is more potent than vitamin D₂. J Clin Endocrinol Metab. 2011.https://pubmed.ncbi.nlm.nih.gov/21177785/ Trang HM, et al. Evidence that vitamin D₃ increases serum 25(OH)D more efficiently. Am J Clin Nutr. 1998.https://pubmed.ncbi.nlm.nih.gov/9701188/ Lehmann U, et al. Bioavailability of vitamin D₂ and D₃. Am J Clin Nutr. 2013.https://pubmed.ncbi.nlm.nih.gov/23616527/ Logan VF, et al. Long-term vitamin D₃ supplementation vs D₂. Br J Nutr. 2013.https://pubmed.ncbi.nlm.nih.gov/23298705/ Jones KS, et al. Ergocalciferol vs cholecalciferol pharmacokinetics. Am J Clin Nutr. 2014.https://pubmed.ncbi.nlm.nih.gov/24695890/ Bouillon R, et al. Vitamin D binding protein and metabolism. Endocr Rev. 2019.https://pubmed.ncbi.nlm.nih.gov/30615155/ Bischoff-Ferrari HA, et al. Effect of vitamin D on falls. JAMA. 2004.https://pubmed.ncbi.nlm.nih.gov/15113819/ Rosen CJ, et al. Nonskeletal effects of vitamin D. Endocr Rev. 2012.https://pubmed.ncbi.nlm.nih.gov/22596255/ Pilz S, et al. Vitamin D and mortality risk. Am J Clin Nutr. 2009.https://pubmed.ncbi.nlm.nih.gov/19116333/ Annweiler C, et al. Vitamin D and cognition. J Intern Med. 2013.https://pubmed.ncbi.nlm.nih.gov/23489360/ Bikle DD. Vitamin D metabolism and clinical implications. Chem Biol. 2014.https://pubmed.ncbi.nlm.nih.gov/24529992/ 👉 Schedule a comprehensive nutritional and metabolic evaluation today. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Bacteria in the Urine: When Not Treating Is Safe—And When It Is Not
Understanding the Risks of Untreated Urinary Infection Introduction The presence of bacteria in the urine— bacteriuria —is one of the most common findings in clinical practice. Yet it is also one of the most misunderstood. A central clinical principle must guide decision-making: Not all bacteria in the urine represent infection—and not all require treatment. At the same time, failing to treat a true urinary infection can carry significant risk, particularly in older adults and medically vulnerable individuals. The challenge is not simply detecting bacteria—it is interpreting what that finding means in the clinical context . Bacteriuria vs. Infection: A Critical Distinction Bacteriuria refers to the presence of bacteria in the urine. It exists in two primary forms: Asymptomatic Bacteriuria (ASB) No urinary symptoms No systemic signs of infection Common in: Older adults Nursing home residents Patients with indwelling catheters Symptomatic Urinary Tract Infection (UTI) Dysuria, urgency, frequency Flank pain or fever Functional decline or confusion (particularly in older adults) Only symptomatic infection generally requires treatment. ¹ Failure to distinguish these entities leads to both overtreatment and undertreatment. When Not Treating Is Appropriate In many patients, particularly older adults, asymptomatic bacteriuria should not be treated . Treatment in these cases: Does not improve outcomes Does not prevent future infection Increases the risk of antibiotic resistance and medication-related complications¹ This principle is well established and widely supported in clinical guidelines. The Clinical Reality: Physician Judgment Is Essential Ultimately, the decision to treat bacteriuria is a clinical determination made by the physician , based on the patient’s symptoms, risk profile, and overall presentation—not on a laboratory result in isolation. While there are well-established scenarios in which bacteriuria may be safely observed, this judgment requires careful evaluation. In many cases—particularly in older adults or medically vulnerable patients—the risk of failing to treat a true infection outweighs the risks associated with appropriate antibiotic therapy . Untreated infection may progress insidiously to pyelonephritis, bacteremia, or sepsis, often with subtle early signs. Clinical context—not the urine test alone—must drive decision-making. Risks of Untreated Urinary Infection and Urosepsis Risks of Not Treating True Infection When bacteriuria represents a true infection , failure to treat can lead to significant complications. 1. Progression to Kidney Infection (Pyelonephritis) Untreated lower urinary infection may ascend: Bladder → kidneys Resulting in: Fever Flank pain Renal inflammation In older adults, early symptoms may be minimal or absent. 2. Bloodstream Infection (Bacteremia) Bacteria may enter the bloodstream from the urinary tract, particularly in: Frail elderly patients Immunocompromised individuals Patients with urinary obstruction This transition significantly increases morbidity and mortality. 3. Sepsis and Septic Shock The most serious consequence: Untreated UTI → Urosepsis → Organ dysfunction → Death ² Clinical features may include: Confusion or delirium Hypotension Rapid breathing Renal dysfunction Metabolic instability In older adults, confusion alone may be the first sign . 4. Functional and Cognitive Decline Even before overt sepsis, untreated infection may lead to: Loss of mobility Increased fall risk Cognitive deterioration Loss of independence These effects can be prolonged and sometimes irreversible. 5. Recurrent or Persistent Infection Failure to treat a true infection may result in: Chronic bacterial reservoirs Recurrent symptomatic episodes Increasing antimicrobial resistance This is especially relevant in patients with: Incomplete bladder emptying Structural urinary abnormalities Biofilm-associated infections Why Infection Is Sometimes Missed Undertreatment often results from diagnostic limitations : Over-reliance on urine dipsticks False-negative nitrite testing Atypical presentations in older adults Delayed or inconclusive cultures A negative screening test does not reliably exclude infection in a high-risk patient. When to Treat vs Observe Bacteriuria Clinical Framework A Practical Clinical Approach Treat when: Urinary symptoms are present Systemic signs exist There is strong clinical suspicion The patient is high-risk Observe when: No symptoms are present No systemic illness is identified Findings are incidental Use advanced diagnostics when needed: Persistent symptoms with negative testing Recurrent infections Atypical presentations Special Situations Where Treatment Is Required Even without symptoms, bacteriuria must be treated in: Pregnancy Before urologic procedures involving mucosal disruption ¹ Bottom Line Bacteria in the urine is not a diagnosis—it is a finding. Overtreatment exposes patients to unnecessary harm Undertreatment allows progression of disease The risk lies not in the bacteria—but in misinterpreting what they represent Failure to treat true urinary infection can result in: Kidney infection Bloodstream infection Sepsis Functional decline Death The goal is not to treat every abnormal test—but to treat the right patient, at the right time, based on sound clinical judgment . Call to Action If you or a loved one has: Recurrent urinary findings Confusion without clear cause Persistent symptoms despite “normal” testing Concern for infection or early sepsis A more thoughtful, clinically guided evaluation may be necessary. At Stages of Life Medical Institute , we emphasize accurate diagnosis, individualized care, and appropriate use of advanced diagnostics to ensure that infection is neither overlooked nor overtreated. 👉 Become a Patient: https://stagesoflifemedicalinstitute.com References Nicolle LE, Gupta K, Bradley SF, et al. Clinical practice guideline for the management of asymptomatic bacteriuria. Clin Infect Dis . 2019;68(10):e83–e110. https://pubmed.ncbi.nlm.nih.gov/30895288/ Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA . 2016;315(8):801–810. https://pubmed.ncbi.nlm.nih.gov/26903338/ The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com
- Pectoralis Minor Syndrome: An Overlooked Cause of Shoulder, Arm, and Nerve Pain
Pectoralis Minor Attaches on the Scapula (Shoulder Blade) and Attaches on the Upper Ribs Introduction Many patients present with arm pain, numbness, or weakness and are told the problem is in the neck, shoulder, or wrist. While those diagnoses are common, there is another frequently missed cause: pectoralis minor syndrome (PMS) . This condition occurs when a small chest muscle compresses nerves and blood vessels traveling into the arm. The good news is that it is highly treatable once identified .¹ What Is Pectoralis Minor Syndrome? Pectoralis minor syndrome is a form of nerve and vascular compression that occurs beneath the pectoralis minor muscle , located in the upper chest. This muscle sits directly over: The brachial plexus The axillary artery The axillary vein When the muscle becomes tight or shortened, it can press on these structures and produce symptoms in the shoulder, arm, and hand.² Anatomy and Location of Compression Pectoralis Minor Anatomy Showing Nerve and Vessel Compression The pectoralis minor attaches from the ribs to a small part of the shoulder blade called the coracoid process . The nerves and blood vessels pass directly underneath this muscle, making it a natural point of compression. Why Does This Condition Develop? In most cases, the cause is posture and muscle imbalance . Common contributors include: Rounded shoulders Forward head posture Prolonged sitting (desk work, driving) Weak upper back muscles Repetitive overhead activity Over time, the chest tightens and pulls the shoulders forward, narrowing the space under the muscle.³ Symptoms to Watch For Nerve-Related Symptoms (Most Common) Pain in the front of the chest or shoulder Tingling or numbness in the arm or hand Weakness or fatigue in the arm Symptoms worse with overhead activity Less Common Findings Arm swelling or heaviness Color changes in the hand Cold sensitivity A key pattern:👉 Symptoms often start in the front of the shoulder/chest and travel down the arm How Is It Diagnosed? 1. Physical Examination The most important step. Findings often include: Tenderness near the front of the shoulder Reproduction of symptoms with arm movement Visible forward shoulder posture 2. Diagnostic Injection A small injection into the pectoralis minor: If symptoms improve → confirms the diagnosis² 3. Imaging (When Needed) Used mainly to rule out other conditions: MRI Ultrasound Treatment: Step-by-Step Approach 1. Posture Correction (Foundation) The goal is simple: 👉 Bring the shoulders back and open the chest Small daily changes can make a large difference: Adjust desk height Raise screens to eye level Avoid prolonged slouching 2. Stretching and Strengthening Pectoralis Minor Stretch and Posture Correction Exercises Stretch the Chest Daily Doorway stretch Corner stretch Strengthen the Upper Back Shoulder blade squeezes Resistance band rows Wall slides 👉 The goal is balance: loosen the front, strengthen the back 3. Hands-On Therapy Massage Myofascial release Trigger point therapy These treatments help relax the tight muscle. 4. Injection Therapy The Green Arrow shows where the medication is injected Options include: Local anesthetic ± anti-inflammatory medication Botulinum toxin (in select, very rare cases) These can: Confirm the diagnosis Provide meaningful symptom relief⁴ 5. Surgery (Uncommon) Reserved for persistent, severe cases: Release of the pectoralis minor tendon Almost never is surgery necessary. Most patients improve without surgery . Common Misdiagnoses This condition is frequently mistaken for: Cervical radiculopathy Rotator cuff disease Carpal tunnel syndrome Recognizing the location and pattern of symptoms is key to avoiding unnecessary treatments. Bottom Line Pectoralis minor syndrome is a common but often overlooked cause of arm pain, numbness, and weakness . It is usually driven by posture and muscle imbalance—and in most cases, it responds well to: Stretching Strengthening Postural correction When needed, targeted injections can both confirm the diagnosis and accelerate recovery. Call to Action If you are experiencing persistent shoulder, arm, or nerve-related symptoms, a careful evaluation can identify whether pectoralis minor syndrome is contributing to your condition. At Stages of Life Medical Institute , we focus on precise diagnosis and targeted treatment to help you return to normal function. 👉 Become a patient: https://www.stagesoflifemedicalinstitute.com References Sanders RJ, Annest SJ. Thoracic outlet and pectoralis minor syndromes. Semin Vasc Surg. 2014;27(2):86–117. https://pubmed.ncbi.nlm.nih.gov/25458043/ Sanders RJ, Hammond SL. Diagnosis of pectoralis minor syndrome. J Vasc Surg. 2007;46(3):601–604. https://pubmed.ncbi.nlm.nih.gov/17826243/ Borstad JD. Resting position variables at the shoulder: evidence to support a posture-impairment association. Phys Ther. 2006;86(4):549–557. https://pubmed.ncbi.nlm.nih.gov/16579673/ Illig KA, Donahue D, Duncan A, et al. Reporting standards of thoracic outlet syndrome. J Vasc Surg. 2016;64(3):e23–e35. https://pubmed.ncbi.nlm.nih.gov/27565607/ The medical references cited in this article are provided for educational purposes only and are intended to support general scientific discussion. They are not a substitute for individualized medical advice, diagnosis, or treatment. Clinical decisions should always be made in consultation with a qualified healthcare professional who can account for a patient’s unique medical history, medications, and circumstances. Subscribe to our Blog Highest Quality, GMP Manufactured Products 1917 Boothe Circle, Suite 171 Longwood, Florida 32750 Tel: 407-679-3337 Fax: 407-678-7246 www.suffernomore.com